
taş adam sendromu (FOP); Tendonlar ve iskelet kasları gibi vücudun normalde kemik olmayan bölgelerinde anormal kemik büyümesi ile karakterize çok nadir görülen kalıtsal bir bağ dokusu hastalığıdır. Bu hastalık özellikle iskelet kaslarının ve yumuşak bağ dokularının kemiğe dönüşmesine neden olur. Eklem bölgelerindeki kemikleşme hareketi zorlaştırır hatta imkansız hale getirir. FOP, doğumdan hemen sonra ayak başparmaklarının şekil bozukluğu ile karakterizedir. Boyun ve kalçaların diğer iskelet anormallikleri.
FOP semptomları erken çocukluk döneminde ortaya çıkar ve yaşam boyu ilerler. Çoğu FOP vakası daha sonraki bir mutasyondan kaynaklanır. Bu hastalığa neden olan genetik mutasyonun vücutta kemik oluşumunu sağlayan bir yolakta BMP adlı bir gende olduğu tespit edilmiştir. BMP sinyal yolu, rahimde fetal iskeletin oluşumu ve doğumdan sonra bebeğin iskelet onarımı için önemlidir.
İçindekiler
belirtiler
FOP’lu tüm insanların anormal ayak parmakları vardır. Bu iskelet değişiklikleri doğumda ortaya çıkar ve bu hastalığın ilk klinik belirtisidir. FOP ile ilişkili en yaygın deformiteler, anormal derecede kısa parmak deformiteleri ve eklem deformiteleridir.
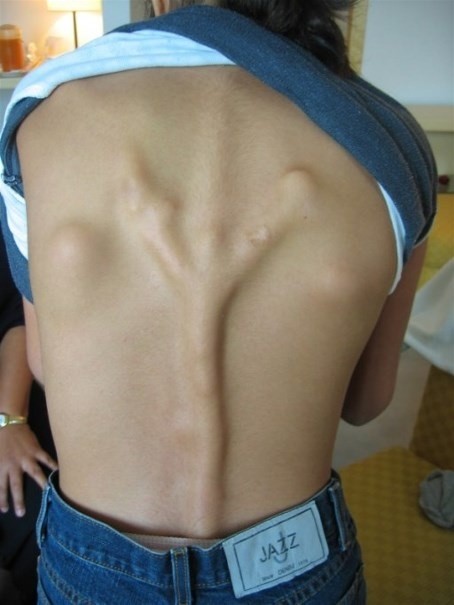
Anormal kemik oluşumu genellikle vücudun erken çocukluk döneminde normalde kemik olmayan yerlerinde meydana gelir. Bu kemikleşme ergenlik, yetişkinlik ve hastaların yaşamları boyunca devam eder. Anormal kemik büyümesi, ilgili bir gende kalıtsal olmayan ve müteakip bir mutasyonun sonucu olarak kendiliğinden ortaya çıkabilir, ancak genellikle bir viral hastalığı veya yumuşak doku yaralanmasını takip eder. Kemikleşmenin ilk belirtisi vücudun yumuşak dokularının, özellikle sırt, boyun ve omuzların şişmesidir. Yumuşak doku şişlikleri, kemiği oluşturan yol boyunca meydana gelir. Genellikle bir kemik büyümesidir; Tendonları, iskelet kaslarını ve bağ dokusunu içerir. Bazı durumlarda bu bölgede ağrı ve sertlik oluşur. Sonunda şişlik azalır ve dokular kemik oluşturmak için sertleşir.
Etkilenen bölgelerde, kemik yavaş yavaş bağ dokusunun yerini alır. İskelet kasına ek olarak kemik büyümesi. Tendon ve bağlarda oluşur. Boyun, sırt, göğüs, kollar ve bacaklar ilk etkilenen yerlerdir. Hastalık son aşamada. Kalçaları, eklemleri, bilekleri, dirsekleri, omuzları ve çeneyi etkileyebilir. Etkilenen bazı bireylerde kemik oluşumu çok hızlı olurken, diğerlerinde süreç kademeli olabilir. Tek yumurta ikizleri arasında bile bu hastalığın ilerleme hızı farklı olabilir.
Vücudun çeşitli bölümlerinin kronik şişmesi, FOP’un ortak bir fiziksel özelliğidir. Anormal kemik büyümesi, etkilenen eklemlerde sertliğe ve hareket kısıtlamasına neden olur. Çene etkilendiğinde hasta konuşmakta ve yemek yemekte zorluk çeker. Anormal kemik büyümesi de omurgada skolyoz oluşumuna yol açabilir. Hastalık ilerledikçe denge, yürüme ve oturma ile ilgili ciddi zorluklar ortaya çıkar.
Son aşamada, FOP hastanın tamamen hareketsiz kalmasına yol açabilir. Hastalar etkilenen bölgelerde sürekli ağrı ve sertlik hissedebilirler. Bir kişinin hareketi kısıtlandığında, solunum problemleri ve kalp yetmezliği meydana gelebilir.
hastalığın nedenleri
2006 yılında Pennsylvania Üniversitesi’ndeki araştırmacılar, BMP yolundaki reseptörlerden birindeki bir mutasyonun FOP’a neden olduğunu buldular. Kemik oluşumu ile ilgili proteinler, fetal oluşumda yer alan düzenleyici proteinlerdir ve doğum sonrası iskelet onarımından sorumludur. FOP ile ilişkili bir gen, ACVR1 adı verilen bir reseptörü kodlar. BMP reseptörleri, kök hücrelerin farklılaşacağı hücre tiplerini belirlemeye yardımcı olur.
İnsan hücrelerinin çekirdeğinde bulunan kromozom, her bireyin genetik bilgisini taşır. İnsan hücreleri normalde 46 kromozom taşır. İnsan kromozom çiftleri 1’den 22’ye kadar numaralandırılmıştır. Cinsiyet kromozomları da X ve Y olarak tanımlanır. Erkeklerde 1 X ve bir Y kromozomu bulunurken, dişilerde iki X kromozomu bulunur. FOP sendromu, gendeki bir mutasyondan kaynaklanır. kromozom 2
Anne ve babadan kalıtılan kromozomlarda da genler bulunur. Anormal bir genin bir kopyası, otozomal dominant hastalıklara neden olmak için yeterlidir. FOP sendromu da kalıtsaldır ve bu durumda ebeveynlerden birinin hasta olması durumunda doğmamış çocuğu etkileme riski %50’dir.
insidans oranı
FOP sendromunun varlığı ilk olarak on yedinci yüzyılda keşfedildi. Dünya çapında 3.000 kişinin bu sendroma sahip olduğu tahmin edilmektedir. Bu sendrom dünyadaki tüm ırkları ve cinsiyetleri etkileyebilir.
FOP teşhis edilirken yanlış teşhis konması yaygındır. Çünkü sadece kısa ve büyük ayak parmaklarına bakılarak tanı konur ancak bu durumdan kaçınılmalıdır. kişileştirme Klinik değerlendirmenin yanı sıra ACVR1 geninde bir mutasyonun varlığının saptanması ile de teşhis konulabilir.
Tedavi Yöntemleri
FOP için kesin bir tedavi yoktur. Bazı ilaçlar ağrı ve şişlik semptomlarını hafifletmek için kullanılabilir. Genetik danışmanlık hizmetleri, çocuk sahibi olmayı düşünen aileler için, çocuğun hastalığa yakalanma riskinin belirlenmesi açısından faydalıdır. Bununla birlikte, FOP’a neden olan mutasyonu hedef alan tedavi modaliteleri ile ilgili çalışmalar devam etmektedir. deneysel tedavi yöntemleri. Antianjiyojenik ajanlar (dokularda kan damarı oluşumunu önleyen ilaçlar) ve BMP antagonistlerini içerebilirler. Kortikosteroidlerin, cox-2 inhibitörlerinin, lökotrien inhibitörlerinin ve talidomidin FOP üzerindeki etkileri de araştırılmaktadır.
kaynak:
https://rarediseases.org/rare-diseases/fibrodysplasia-ossificans-progressiva/
yazar: Ayka Olkay
Diğer gönderilerimize göz at
[wpcin-random-posts]